Research Overview
Our current interesest and potential directions are as below (but will be transforming over time!).
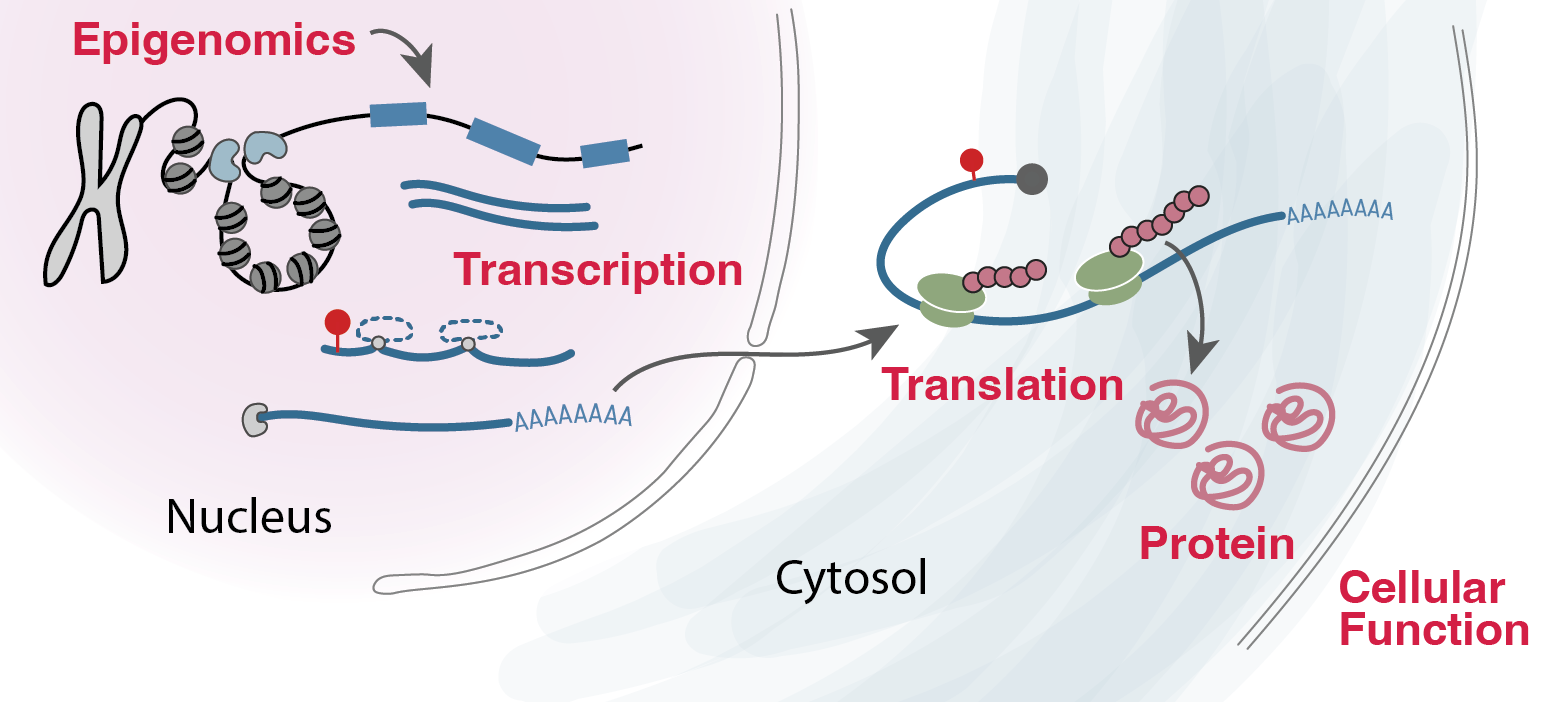
Functional characterization of disease alleles in time⌛ and space🪐
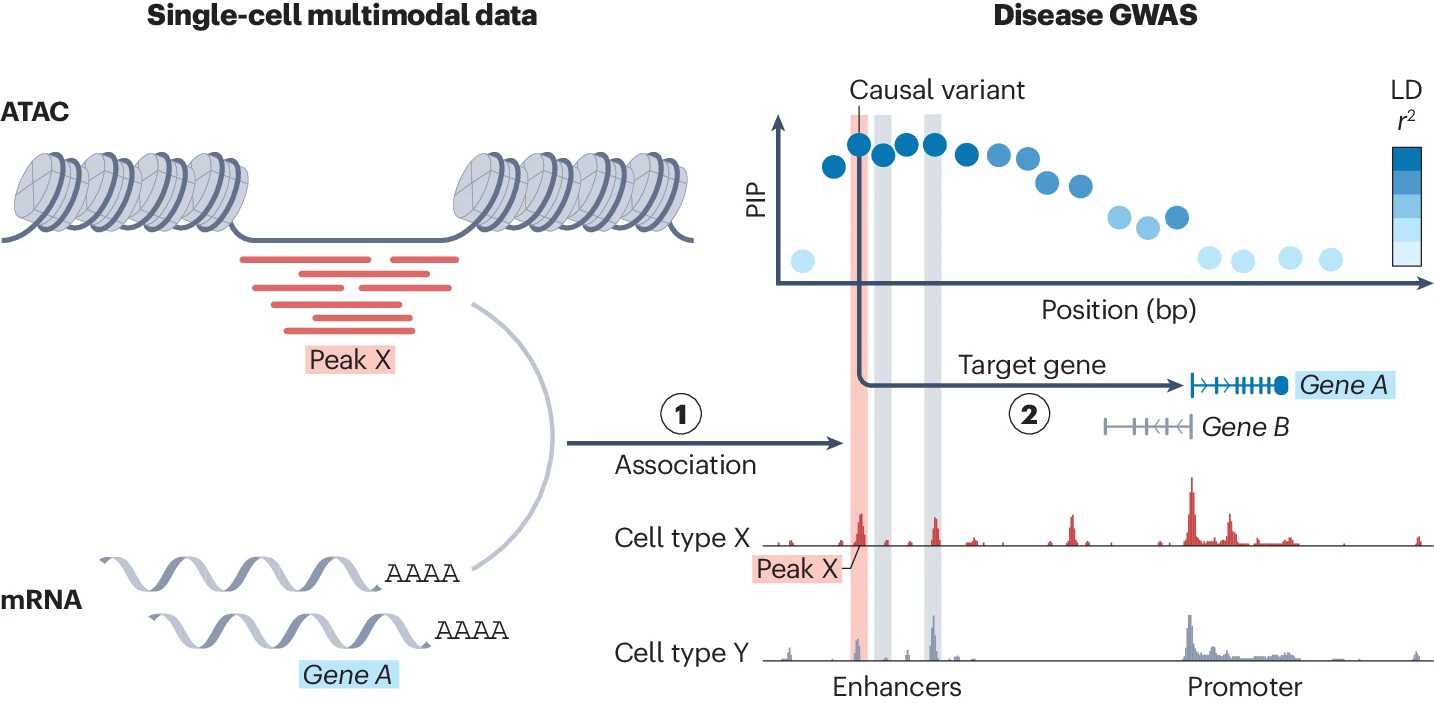
More than 90% of common disease loci act through non-coding mechanisms, in which risk variants modulate disease risk by altering gene expression abundance but not protein function. Even in rare diseases, the coding function has not yet sufficiently explained ~50% of causal mechanisms in patients, leaving the majority of patients with uncertain diagnoses and suboptimal therapeutics. Defining non-coding variants to their function in gene regulation and cellular phenotype is a huge open question in the field.
We are interested in addressing this question by developing statistical methods that integrate various molecular recordings in the cells and identify which cells, when in our life, and where in our body these variants influence gene regulation. We’ve already learned that these regulatory changes are highly context-specific, and so are interested in using molecular profiling (chromatin, RNA or post-transcriptional modifications and translation?) from various aspects in our cells in time and space through collaborations with experimentalists.
Relevant publications
- Sakaue S et al. Tissue-specific enhancer-gene maps from multimodal single-cell data identify causal disease alleles. Nat Genet. 2024 Apr; 56(4):615-626. PMID: 38594305; PMCID: PMC11456345.
- Sakaue S. SCENT defines non-coding disease mechanisms using single-cell multi-omics. Nat Rev Genet. 2024 Sep; 25(9):597. PMID: 38816646.
- Sakaue S & Raychaudhuri S. Early and late RNA eQTL are driven by different genetic mechanisms. bioRxiv. 2025 Feb.
Decoding complex genomic regions🧬 with new sequencing technologies
While recent sequencing technology has largely revealed genetic variations among us, we still lack a comprehensive understanding of very complex genome regions. Repetitive, duplicated, or hyperpolymorphic regions were such examples, including the HLA and KIR regions. But there is a reason why they are so complicated - because they are important for our biological function such as immunity and under strong selective pressure. For example, HLA genes confer the largest number of disease associations of any locus genome-wide, with strongest effects on autoimmune diseases.
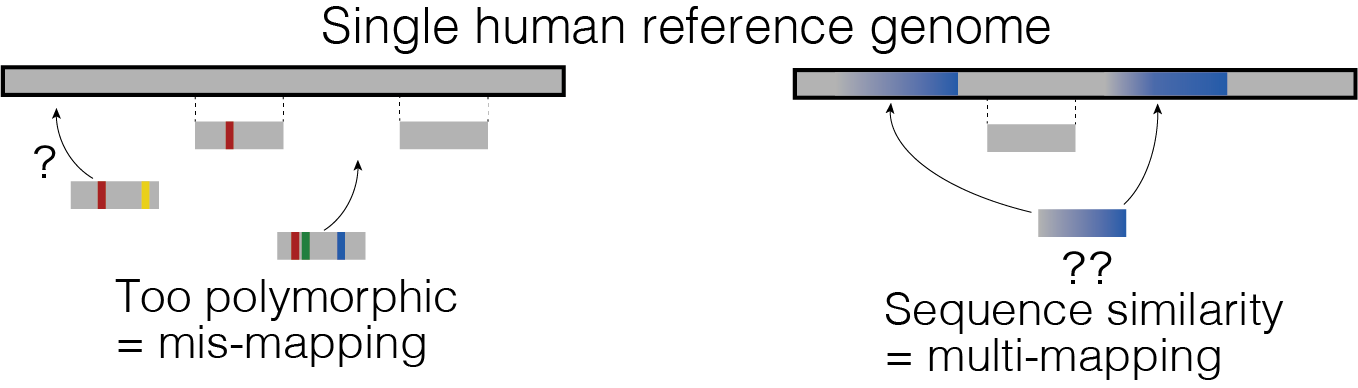
We are interested in developing statistical methods to accurately decode these complex regions by using data from new sequencing technologies (such as in T2T project) and identify yet-to-be-seen important variations in this dark matter in the genome that might play a role in human traits and diseases.
Relevant publications
- Sakaue S, et al. Tutorial: a statistical genetics guide to identifying HLA alleles driving complex disease. Nat Protoc. 2023 09; 18(9):2625-2641. PMID: 37495751; PMCID: PMC10786448.
- Kang J, … Sakaue S, Raychaudhuri S. Mapping the dynamic genetic regulatory architecture of HLA genes at single-cell resolution. Nat Genet . 2023 Dec;55(12):2255-2268. doi: 10.1038/s41588-023-01586-6. PMID: 38036787; PMCID: PMC10787945.
- Sakaue S, et al. Decoding the diversity of killer immunoglobulin-like receptor by deep target sequencing and a high-resolution imputation method. Cell Genomics 2(3) 100101-100101 (2022).

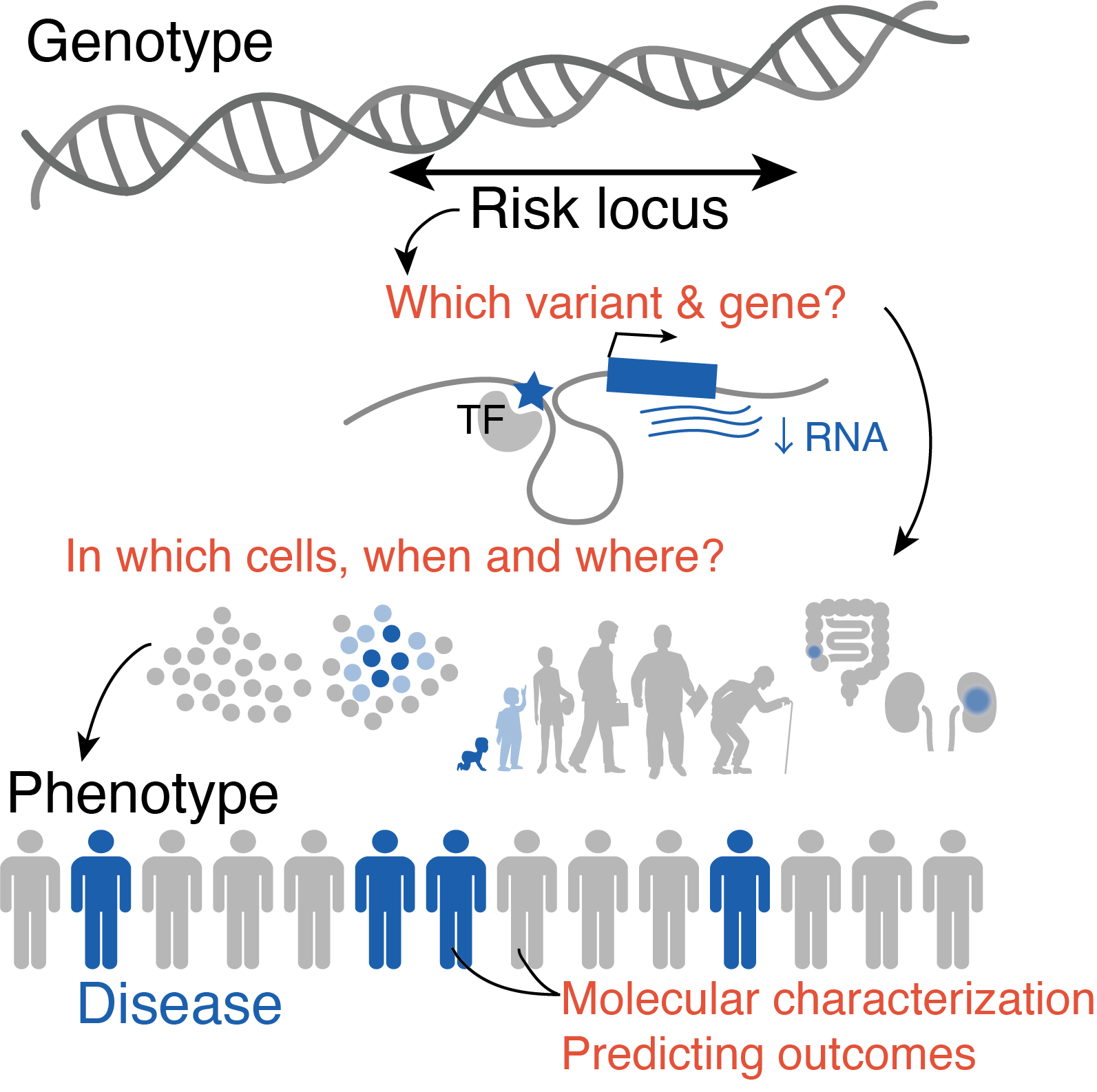
How can we translate our biological understanding of genomic risks into clinically actionable insights?
With these great genomic technologies and large-scale datasets in our hands, we are yet to be fully leveraging our genomic understanding into clinical practice. For example, currently used disease entities often include heterogenous disease conditions that could delay the delivery of most effective treatment. These disease entities have oftentimes been constructed from empirical clinical knowledge but not informed of molecular profiling of the patients.
While genetics has been conventionally focused on identifying risk alleles or molecules associated with a predefined disease category, we could use genetics for data-driven disease definitions. Genetics as a tool is ideal in this context, since genetic variation always precedes development of disease conditions, thereby suggesting causal relationships and utility in predicting outcomes. We could further identify data-driven disease subtypes by using high-dimensional genomics modalities, with effective statistical models. We can anticipate that the significant associations will point us towards key genetic and molecular features for classifying patients into fine-grained disease subtypes, that may help patients reach to most effective treatment strategies without trials and errors.
Relevant publications
- Sakaue S, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021 Oct;53(10):1415-1424. PMID: 34594039
- Sakaue S, et al. Trans-biobank analysis with 676,000 individuals elucidates the association of polygenic risk scores of complex traits with human lifespan. Nat Med. 2020 Apr;26(4):542-548. PMID: 32251405